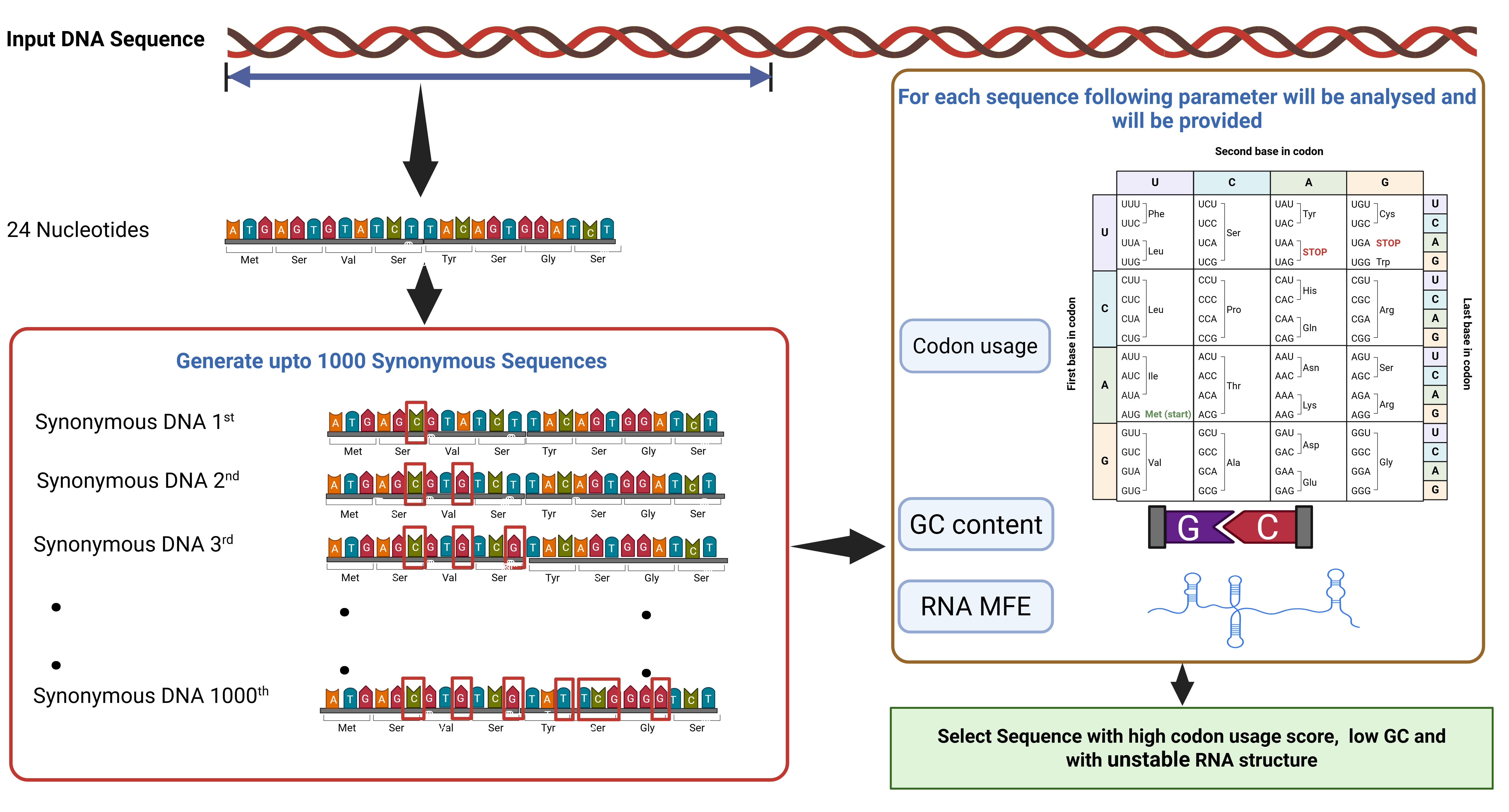
This web server is designed to predict optimal synonymous DNA sequences for heterologous expression in Escherichia coli; the webserver is based on the mRNA secondary structure of the 5′ end minimum free energy (i.e. mRNA fold energy values), codon usage, and GC content.
mRNA Secondary Structure Visualization
×
![]()
Full-size view of mRNA folding visualization
Codon Usage Score
This score is a measure of how frequently a particular codon is used in E. coli. This score is calculated using the codon usage frequency of each codon per thousand nucleotides in E. coli.
GC Content Score
This score is a measure of the percentage of guanine (G) and cytosine (C) nucleotides in a DNA sequence. G and C nucleotides form three hydrogen bonds, while adenine (A) and thymine (T) nucleotides form two hydrogen bonds. As a result, DNA sequences with higher GC content are typically more stable.
RNA Structure
The structure of mRNA at the 5 prime end dictates translational efficiency. An mRNA that forms stable secondary structures is less likely to be translated. It uses the Vienna RNAfold package to calculate the free energy (kcal/mol-1) of the mRNA ensemble. The synonymous sequence that disrupts the stable secondary structure of mRNA is likely to have higher translational efficiency.
The web server works by entering the DNA sequence as input. Then the web server will estimate the values of these three parameters for the entered sequence and provide all the possible synonymous sequences. The user can use this information to select sequences that are likely to be optimally translated in E. coli